|
琉球大学医学部の藤本真悟技術補佐員、琉球大学大学院医学研究科の木村亮介教授、静岡県立大学の明正大純助教らの研究チームによる研究成果が、オープンアクセスの学際的電子ジャーナル「Scientific Reports」誌に掲載されました。本件に関する取材については、下記のとおりになりますので、よろしくお願いします。 <発表のポイント> ◆日本のメダカ類2種の集団構造と交雑を分析して、東日本太平洋側に分布するミナミメダカO. latipes集団が日本海側に分布するキタノメダカO. sakaizumiiと過去に交雑したことを示しました。また、飼育品種ヒメダカの核遺伝子の由来も評価し、詳細な過程を新たに明らかにしました。 ◆本研究が提案する核ゲノムの一塩基変異(SNVs)の遺伝子型決定および交雑分析の手法は、メダカの放流による野生集団への遺伝的影響や野生動物の集団遺伝構造を評価するのに貢献します。
|
<発表概要>
PCR産物とショートリードシーケンサー*1を用いてSNVs(核ゲノムの一塩基変異)を簡便に決定する分析技術を適用して、日本のメダカ類の集団遺伝構造と交雑を分析しました。メダカ類は主に太平洋側に分布するミナミメダカOryzias latipesと日本海側に分布するキタノメダカO. sakaizumiiの2種に分化しています。本研究で、初めて東北地方でもそれらが過去に交雑した可能性を示しました。また、広く販売されている黄色変異品種ヒメダカのSNVsを決定して、瀬戸内海周辺と東日本に広域分布する2つのミナミメダカ野生集団が核遺伝子の由来に寄与することを確認しました。ヒメダカは人為的な放流で野生集団に遺伝的撹乱*2をもたらすことが懸念されています。本研究が提案した交雑分析手法は、放流の遺伝的影響を評価することに貢献します。
①研究の背景と目的
1)PCR産物を用いるシーケンス技術の出力改善
SNVsを用いて個体の遺伝的組成を分析することで、過去に起きた集団間の分化や交雑といった集団の成り立ちを推定できます。こうした知見は、私達ヒトの歴史の復元にも用いられますが、野生動物の進化や生態を理解する上でも基礎となる知見です。SNVsを解読する手法はいくつかありますが、PCR(Polymerase Chain Reaction)法で得られた産物を配列決定する方法は、少量のDNAで分析できる利点があります。近年、3~10塩基の少数種類の塩基配列を用いて、ゲノムの多数箇所の塩基配列を一度に決定する手法*3がいくつか提案されていました。本研究ではこれらを、MAAS(multiple arbitrary amplicon sequencing)と総称します。MAASの先行事例の多くは数10~100個のSNVsを得て解析しました。しかしながら、過去の集団間での交雑を推定する場合、信頼性の高い分析結果を得るためには、SNVsの数をさらに一桁か二桁は増やして改善する必要がありました。本研究はメダカを材料に、MAASの効率改善を試みるとともに、集団遺伝構造や交雑分析の有効性を検証しました。
2)日本産メダカ2種の野生集団の集団遺伝構造と交雑の分析
1980年代から2000年代にかけて竹花らが収集したDNAサンプル*4を再解析して、核ゲノムの集団遺伝構造と交雑を分析しました。西日本の日本海側では、過去に自然下で交雑を経験した集団がいることも知られており、丹波但馬地方に交雑集団が分布します。一方で、東日本に分布する集団は近年の人為的放流に起因すると推測された地点を除いて、2種間の交雑は知られていません。そこで東日本に分布する2種の野生集団間での交雑を検討しました。
3)飼育品種ヒメダカの遺伝的由来
本研究では、さらに黄色の体色を持つ飼育品種ヒメダカも同様の交雑分析手法で評価して、この品種が持つ核ゲノムの遺伝的由来を確認しました。ヒメダカは人為的放流で野生集団に遺伝的撹乱をもたらすと懸念されています。遺伝的組成を明らかにすれば放流の有無を検出して、野生集団への影響を評価できます。
②研究内容
1)MAAS技術の出力改善
一度に多種類の産物(DNAの断片)をPCRで増幅する場合、塩基配列の異なるそれぞれの産物の増幅効率は異なります。こうしたPCR産物で塩基配列を決定すると、増幅されやすい同じ配列ばかり検出するため、多種類の塩基配列を決定する効率が低下します。一様に増幅しやすいプライマー配列(PCR用の試薬のひとつ)を得るため、図1で示す手順で新規プライマーを設計しました。また、サンプルあたりに費やすシーケンスの読取り数と決定された塩基数の関係を評価して、先行して使われているMIG-seq法より効率良く塩基を決定できることを示しました。
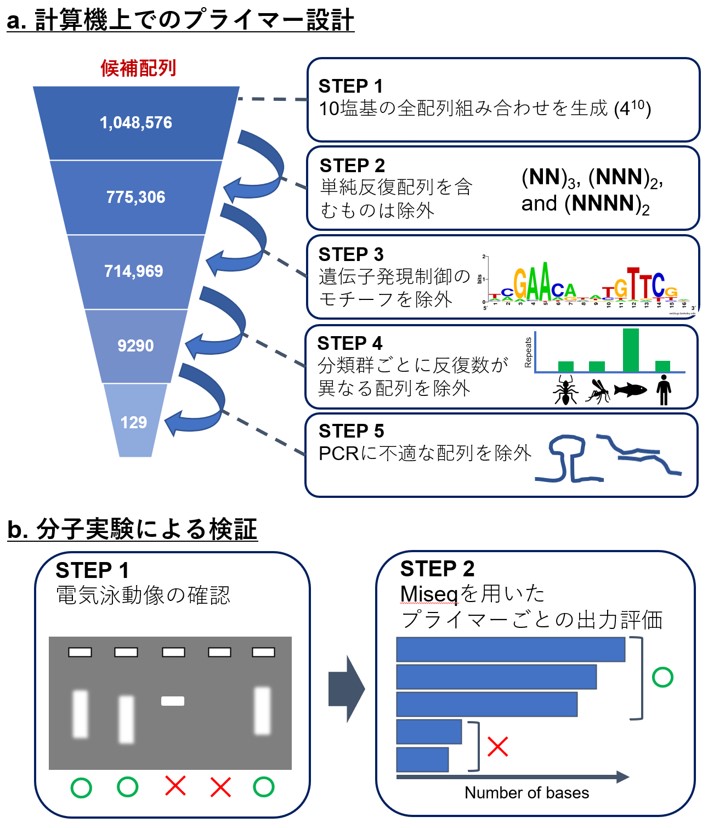
図1. MAASで用いるプライマー配列の特定と検証
2)メダカ野生集団の集団遺伝構造と交雑の分析
2-1. 野生集団および飼育品種の系統関係とADMIXTUREによるグループ分け
新規設計したプライマーを用いたシーケンスで得られた核遺伝子のSNVsで、近隣結合法による系統関係を推定しました。ミナミメダカとキタノメダカの2種は遺伝的に大きく分化し、ミナミメダカの中では、九州の東シナ海側と南西諸島に分布するクレードIとそれ以外の日本全国に分布するクレードIIに分かれました(図2, 3)。これらの結果は、先行研究でミトコンドリアや核遺伝子で調べた系統関係と一致します。
遺伝子組成の類似性でグループ分けを行うADMIXTUREによる分析も同様の傾向を示しました。また、ヒメダカはミナミメダカの中でも、実験系統として確立された近交系*5 Hd-rRと同じグループに位置付けられました(図3)。
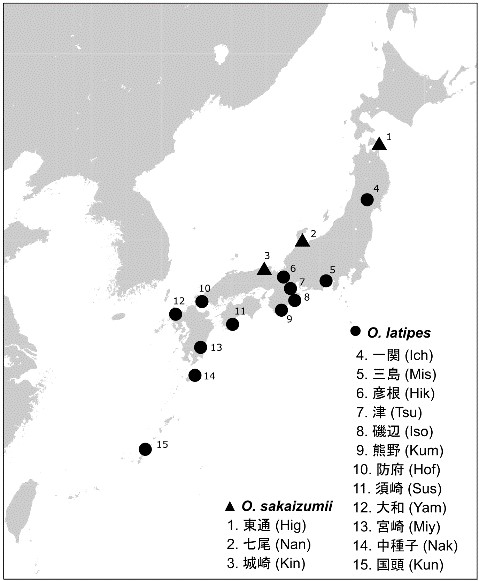
図2. 野生集団の採集地点
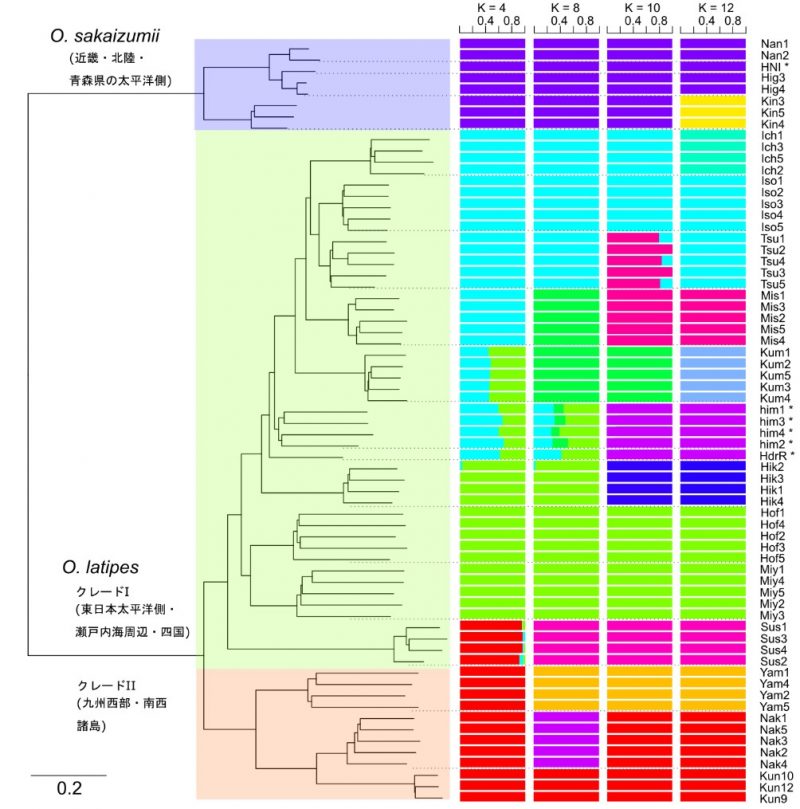
図3. 近隣結合法による系統関係とADMIXTUREによるグループ分け
(各個体のラベルは図2の地点名に対応する。*は飼育系統を表し、ヒメダカ (him)、
キタノメダカ(HNI) およびミナミメダカ(Hd-rR)の近交系を含む)
2-2. 東日本の野生集団およびヒメダカにおける交雑の分析
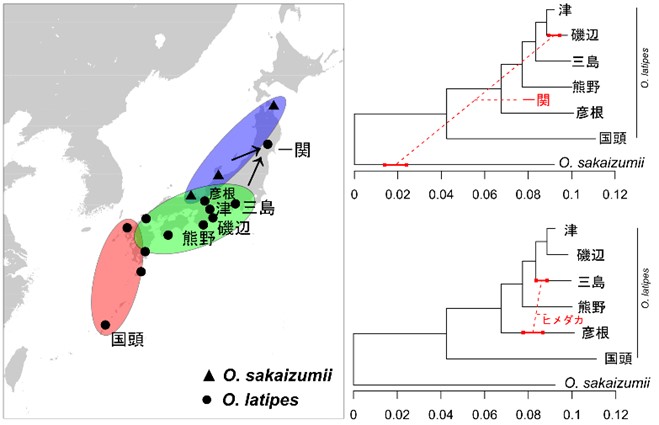
野生集団間での交雑についてさらに分析を進めたところ、岩手県一関から得られたミナミメダカ集団は、キタノメダカと過去に交雑していたことが示唆されました(図4a, c)。紀伊半島より東に位置する東海地方や東北地方の太平洋側は、現在ではミナミメダカのみ分布します。この結果は東北のミナミメダカ集団が形成される過程で過去に2種の交雑があったことを示唆します。この分析では、ヒトの集団遺伝構造と交雑を解析する目的で発展した、ABBA-BABAテストおよびF統計量と呼ばれる分析手法にもとづいて、交雑シナリオを構築しました。
同様の分析を飼育品種のヒメダカにも適用して遺伝的由来を検討したところ、ミナミメダカに含まれる遺伝子型のうち、瀬戸内海周辺の西日本に広く分布する遺伝子型(彦根)と、東海地方から東北地方の東日本に広く分布する遺伝子型(三島)の2集団に近縁な系統が寄与したことが分かりました。この結果はミトコンドリアDNAのハプロタイプで報告された傾向を核遺伝子でも裏付け、さらに詳細なシナリオを推定できたことになります。
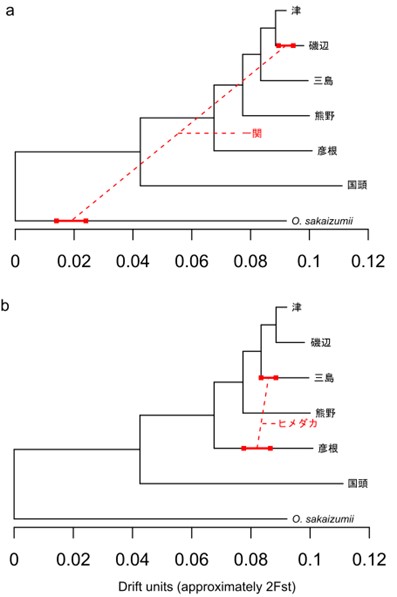 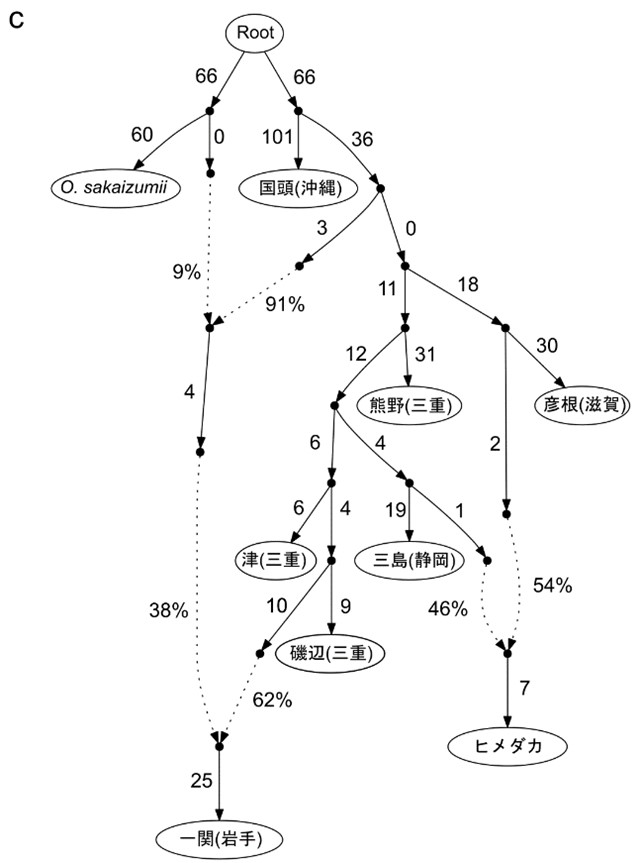 図4. MixMapper(左)および、admixturegraph(右)で推定した交雑シナリオ(実線と数値は遺伝的浮動、破線と割合%は集団間の交雑を示す) |
③ 本研究のまとめと意義
MAASという簡便にSNVsを決定できる分析技術を日本のメダカに適用して2987 SNVsを、67個体から欠損なしで得られました。交雑分析の結果は東日本でミナミメダカとキタノメダカの野生集団が交雑したことを示唆しました。野生集団から得られたSNVsの情報は野生集団の保全における地域集団の定義にも役立ちます。さらに、遺伝的撹乱をもたらと懸念されているヒメダカの核遺伝子の由来も明らかにしました。本研究が提案した遺伝子型決定および交雑分析の手法は、生物多様性の保全における野生集団の遺伝構造の評価に貢献します。
<用語解説と脚注>
*1, ショートリードシーケンサー: 次世代シーケンサーとも呼ばれる塩基配列の読み取り機械です。一度の処理で、長くても数百 bpの短い塩基配列を数百万から数十億の本数、並列的に多数決定することができるので、ヒトの全ゲノム解析で広く使われるようになりました。メーカーとしてはIlluminaが最も有名で本研究では、Hiseq Xを用いました。
*2, 遺伝的撹乱: メダカを始めとした多くの生物は、同一種内でも地理的に離れた地域集団間では塩基配列や遺伝子頻度が異なります。ある種が自然条件下で移動不可能な場所へ、人為的な移動で運ばれると交雑による遺伝子の移入が起きます。これは野外に存在しなかった遺伝的パターンを生じるので、進化や生態を研究する上でノイズになります。また、飼育品種はしばしば飼育環境に適した遺伝子型が固定していますが(ヒトが好む 目立つ体色や形態なども含む)、そうした遺伝子型は野生環境で生存に不利になりやすいため、遺伝子の移入を受けた在来集団の存続にも負の影響をあたえることがあります。
*3, MIG-seq (Suyama and Matsuki, 2015, Scientific Reports), RAM-seq (Bayerl et al. 2018, Molecular Ecology Resources), GRAS-Di (トヨタ自動車)など。
*4, 竹花らが収集したDNAサンプル: ミトコンドリアDNAの部分配列を用いてメダカの集団遺伝構造を分析した先行研究(Takehana et al. 2003, Zoological Science)で使われたDNAサンプル。元新潟大学理学部教員の酒泉満博士から本研究に用いるため、共著者の明正大純博士に譲渡された(現在は、静岡県立大学に所蔵)
*5, 近交系: 兄妹・姉弟同士の近親交配を20世代以上継続して得られる、ほとんどすべての遺伝子座がホモ接合になった動植物の系統。HdrRとHNIはそれぞれ、ミナミメダカとキタノメダカのゲノムの参照配列を決定する解析でも用いられた(Ichikawa et al. 2017, Nature Communications)
<謝辞>
分子実験の機器と施設の利用について、琉球大学研究基盤センターに感謝します。本研究は、公益財団法人・河川財団の河川基金(2017-5211-062)、日本学術振興会科研費助成金(JP19K16232)、および琉球大学が推進する「亜熱帯島嶼の時空間ゲノミクスプロジェクト」による支援を受けました
<論文情報>
論文タイトル:Population admixtures in medaka inferred by multiple arbitrary amplicon sequencing(和訳:MAAS法を用いて推定したメダカ野生集団の交雑)
雑誌名:Scientific Reports
著者名:Shingo Fujimoto, Hajime Yaguchi, Taijun Myosho, Hiroaki Aoyama, Yukuto Sato & Ryosuke Kimura(藤本真悟*1、矢口甫*2、明正大純*3、青山洋昭*4、佐藤行人*1、木村亮介*5)
アブストラクトURL: https://www.nature.com/articles/s41598-022-24498-7
DOI:https://doi.org/10.1038/s41598-022-24498-7
著者所属
*1: 琉球大学 医学部附属実験実習機器センター; *2: 関西学院大学 理工学部 生命科学科; *3: 静岡県立大学 食品栄養科学部 環境生命科学科 生態発生遺伝学研究室; *4: 琉球大学 戦略的研究プロジェクトセンター; *5: 琉球大学大学院 医学研究科 人体解剖学講座
