|
脊髄性筋萎縮症(SMA)は、脊髄前角細胞の変性・消失により、筋力低下や筋萎縮が進行する難病です。重症例では生後6か月までに発症し、2歳までに死亡する方がほとんどです。 |
<概要>
琉球大学病院小児科の中西浩一教授、知念安紹診療教授、仲村貞郎診療講師らは2024年2月、沖縄県で初めて、新生児スクリーニング※1により脊髄性筋萎縮症(SMA)であることが判明した新生児において、症状が発現する前(未発症)の遺伝子治療を行いました。
SMAは、運動神経細胞生存遺伝子(SMN遺伝子)※2の異常によってSMNタンパクが十分に作られず、脊髄前角細胞が変性・消失し、筋力低下や筋萎縮を起こす難病です。重症例では人工呼吸器が必要となり、無治療では多くは2歳までに死亡します。従来は治療困難でしたが、近年、この病気に対する遺伝子治療薬が開発されました。この治療を行うと、正常なSMN遺伝子が患者の運動ニューロンで働き、筋力低下や筋萎縮を防ぐことができます。しかし、症状が現れてしまうと治療の効果が乏しく、症状進行は抑えることができても回復は難しいことがわかっています。一方で症状発現前に治療を開始すると、ほぼ正常な発育が認められ完治も期待できます。以上のことから、症状が現れる前にSMA患者を発見することが重要です。
沖縄県では2023年6月からSMAを含む9疾患の新生児オプショナルスクリーニングが開始され、実際にSMA患者を症状の発現前に診断することができました。その結果、遺伝子治療を高い治療効果が期待できる症状の発現前に開始することができました。琉球大学病院では今後も新生児における先天性疾患の早期発見と早期治療に努めてまいります。
<説明>
[背景]
脊髄性筋萎縮症(SMA)は、運動神経細胞生存遺伝子(SMN遺伝子)の異常によってSMNタンパク質が十分に作られず、運動神経細胞である脊髄前角細胞が変性・消失し、筋力の低下や筋の萎縮が進行する国の指定難病の一つです。重症例では人工呼吸器が必要となり、治療を行わない場合、多くは2歳までに死亡します。SMA患者の発症頻度は1万人に1~2人と推定されています。近年、スピンラザ®※3やゾルゲンスマ®※4などの飛躍的に進歩した治療薬が承認され、SMAの治療の選択肢が増えました。その効果は治療を行った時期が早ければ早いほど高いことが分かっています(図1)。すなわち、最大限の治療効果を得るためには、症状の発症前にSMAの患者を見つけて、治療することが望まれます。わが国でも、症状の発現前にSMAの診断がされた兄弟に遺伝子治療が行われ、高い治療効果が得られた報告があります。しかし、家族歴がない場合の早期診断は非常に困難でした。
[内容・成果]
わが国では、生後4日目から6日目の間に、全ての赤ちゃんの血液をろ紙に染み込ませた乾燥ろ紙血を用いた新生児スクリーニング(マススクリーニング)が行われています。マススクリーニングの対象とはなっていないSMAについても、このようなろ紙を用いて新生児スクリーニングができれば理想的です。そこで、家族歴の有無に関わらず患者を早期発見するための方法が開発され、世界的に実施されるようになり、わが国でも急速に普及してきました。
SMAの原因となるSMN遺伝子異常のほとんどは、SMN1という遺伝子の欠失で起こります。すでに、わずかな血液から、そのSMN1遺伝子欠失を定量PCR法※5で判定し、SMAの可能性がある新生児を発見する方法が確立されています。一般社団法人 稀少疾患の医療と研究を推進する会(CReARID [クレアリッド])による新生児スクリーニング(オプショナルスクリーニング)の対象に、SMAが含まれています。
沖縄県では2023年4月に関連5団体(沖縄県医師会・沖縄県産婦人科医会・沖縄産科婦人科学会・沖縄県小児科医会・沖縄小児科学会)により沖縄こども先進医療協議会(Okinawa Children’s Advanced Clinical Research Association [O-CHART])が設立され、クレアリッドとの連携のもと、同年6月からSMAを含む9疾患の新生児オプショナルスクリーニングが開始されました。
この度、2024年1月に生後5日目にお産施設で採血された新生児の試料用いて検査を行ったところ、SMAの可能性のある新生児が生後13日目の時点で発見されました。その後、琉球大学病院小児科で県外専門施設の協力のもと、精査を進めてSMAと診断し、電気生理的検査において未発症を確認し、同年2月に遺伝子治療薬(ゾルゲンスマ®)を投与しました。投与した時点(生後38日目)では明らかなSMAの症状は認めず、症状の発現前に治療を行うことに成功しました。
[展開]
2024年3月現在、わが国でも多くの都道府県でSMAの新生児スクリーニングが行われています。オプショナルスクリーニングは任意かつ有料の検査ですが、一部においては公的資金による補助が開始されています。最終的にはすべて新生児がこのスクリーニング検査を受けられるようになることが期待されます。琉球大学病院ではあらゆるタイプの新生児スクリーニングを活用し、新生児における先天性疾患の早期発見と早期治療に務めてまいります。
[用語解説]
※1 新生児スクリーニング:放置すると将来、障害が出てくるような先天異常症等を生後早期に発見して、症状の発現前に治療介入して障害を予防するシステム。現時点では厚労省の指導の下で自治体が主体となって実施する20疾患を対象としたマススクリーニングと、希望者が選択して受ける9疾患を対象としたオプショナルスクリーニングがある。SMAはオプショナルスクリーニングの対象である9疾患のうちの1つ。
※2 運動神経細胞生存遺伝子(SMN遺伝子):ヒトの5番染色体上にあり、SMN1遺伝子とSMN2遺伝子がある。SMNタンパク質の約90%はSMN1遺伝子から作られ、残りがSMN2遺伝子から作られる。患者の大半は、両親からSMN1遺伝子の欠失を受け継ぐことで、SMAを発症する。またSMN2遺伝子のコピー数が少ないほどSMAの重症度が高くなる傾向にある。
※3 スピンラザ®:一般名ヌシネルセンナトリウム。世界初のSMA治療薬で、わが国では2017年に承認された。定期的に髄腔内に投与することで、運動ニューロンのSMN2遺伝子に働きかけ、SMN2遺伝子から作られるSMNタンパク質が増加し、生命予後および運動機能の改善が期待できるが、症状が出る前に投与することが重要と考えられている。
※4 ゾルゲンスマ®:一般名オナセムノゲンアベパルボベク。アデノ随伴ウイルスを利用し、正常なSMN遺伝子を運動ニューロンへ導入することで安定的にSMNタンパク質を発現することができる遺伝子治療用ベクター製品。1回の静脈内点滴投与で生命予後および運動機能の改善が期待できるが、症状が出る前に投与することが重要と考えられている。
※5 定量PCR法:目的とする遺伝子をPCR(ポリメラーゼ連鎖反応)による増幅反応をリアルタイムにモニタリングして測定する方法。大半のSMA患者ではSMN1遺伝子欠失のホモ接合体のために、SMN1遺伝子のDNAは増幅されないと考えられる。
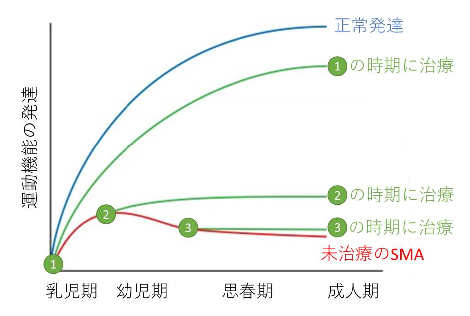
図1 治療時期と治療効果の関係を模式化した図。青い線は正常の運動発達、赤い線は未治療のSMA患者の運動発達、緑の線は治療をしたSMA患者の運動発達を示している。数字は治療時期を示し、1、2、3の順に治療する時期が早いほど、運動機能の発達が良く治療効果が高い。(Sumner CJ, et al. J Clin Invest. 2018より改変)
